随着人们对抗生素的滥用越来越严重,细菌在自然选择的压力下,在与抗生素的斗争中逐渐掌握了逃避抗生素攻击的机制,于是越来越多具有耐药性的变异细菌菌株陆续被发现,致病细菌再次卷土重来,迅速进化出对抗生素耐药的能力,抗生素对细菌的封锁线面临坍塌,使得细菌耐药性逐渐成为人类健康安全最大的威胁之一。
近年来,一类合成的抗生素-恶唑烷酮类抗生素,其对耐药菌,如耐甲氧西林的金黄色葡萄球菌(MRSA)、耐甲氧西林表皮葡萄球菌(MRSE)、抗万古霉素肠球菌(VRE)等展现出优异的抗菌活性,抑制细菌蛋白质合成的初始阶段来发挥抗菌作用且与其他抗生素无交叉耐药性,凭借这些出色的特性吸引到了越来越多研究者的关注。2000年,首个恶唑烷酮类抗生素利奈唑胺在美国FDA批准上市用于治疗皮肤、组织感染、细菌性肺炎以及其他感染性疾病。利奈唑胺在治疗的初期阶段显示出令人满意的治疗效果,但随着长期的使用暴露出骨髓抑制、血小板减少等不良反应并且出现了利奈唑胺的耐药性细菌,这些问题促使着药物化学家们对其结构进行不断地优化以获得具有改善的安全性以及更高抗菌活性的恶唑烷酮类抗菌剂。
陕西科技大学梁承远课题组基于对恶唑烷酮类抗菌剂的研究与兴趣,对过去十年研究者们在改善这类抗生素的安全性和药代动力学特征,提高这类抗生素的抗菌活性上所做的工作进行了系统的总结、讨论和展望。此外,还对两种已上市的恶唑烷酮类抗生素(利奈唑胺和泰地唑胺)目前正在进行的研究以及含有恶唑烷酮支架的新型抗菌剂进行了总结。

图1.含恶唑烷酮支架抗菌药物的研究现状和展望。
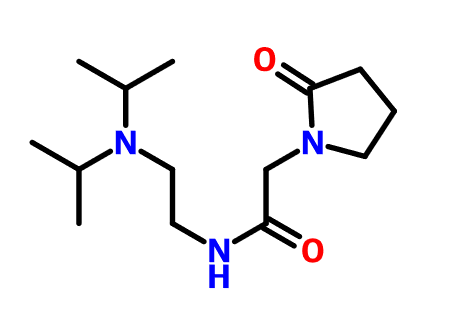
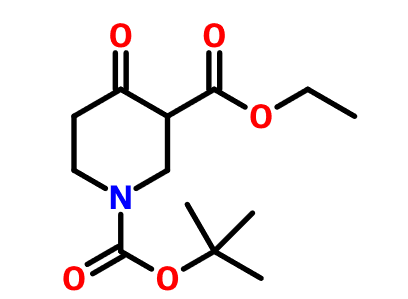
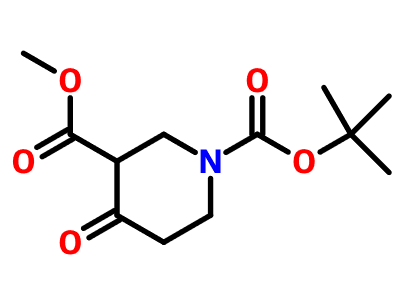
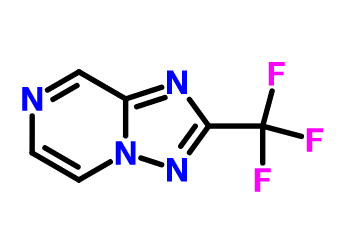
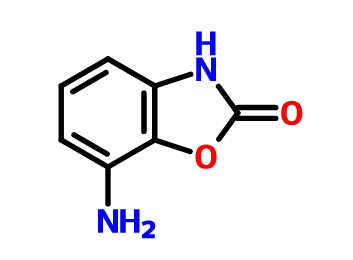
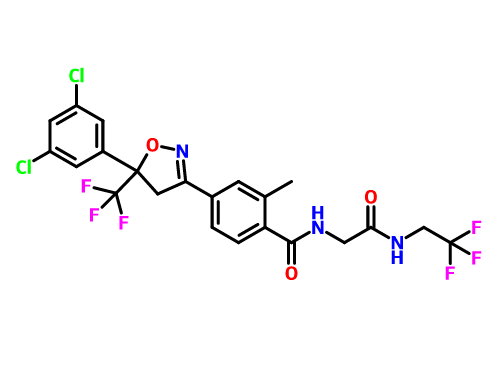
目前对于恶唑烷酮类抗菌剂的研究主要集中在基于利奈唑胺的骨架进行修饰,包括末端的吗啉环修饰、C5位侧链的改造以及中心三环融合骨架的构造。构效关系总结表明,使用[1,2,5]三氮己烷、二氢吡啶酮、吡唑并[1,5-a]吡啶等药效基团取代利奈唑胺末端的吗啉基所得到的恶唑烷酮类抗菌剂具有更强的抗菌活性并且显示出较低的骨髓抑制作用和单胺氧化酶的抑制活性,此外,中心三环融合骨架的构建进一步提高了化合物的可成药性,例如末端含有硫代吗啉基团和C头部含有1,2,3-三唑侧链的三环融合类恶唑烷酮抗菌剂不仅便显出优异的抗菌活性、改善的安全性,还显示出高水溶性、良好的PK和口服生物利用度、优异的代谢稳定性、低细胞毒性和CYP450酶抑制等可成药性特征,这些结果使得中心具有三环融合骨架的恶唑烷酮类抗菌剂成为流行的研究方向。一些新型的含恶唑烷酮基团的抗菌剂如基于LpxC抑制剂改造恶唑烷酮类衍生物,也显示出良好的抗菌性能和广谱的抗菌活性,这些都值得进行进一步的研究。

图2. 代表性的基于利奈唑胺改造得到的恶唑烷酮类衍生物和新型的恶唑烷酮类衍生物。图片来源 J. Med. Chem.
这一成果近期发表在Journal of Medicinal Chemistry上,文章的第一作者是陕西科技大学赵倩倩讲师,通讯作者是梁承远副教授。
论文信息:
https://pubs.acs.org/doi/full/10.1021/acs.jmedchem.1c00480
Current Landscape and Future Perspective of Oxazolidinone ScaffoldsContaining Antibacterial Drugs. Qianqian Zhao, Liang Xin, Yuzhi Liu, Chengyuan Liang *, Jingyi Li, YanlinJian, Han Li, Zhenfeng Shi, Hong Liu, Wenqiang Cao. J. Med. Chem. 2021
梁承远博士简介
梁承远,陕西科技大学药学系副教授,陕西省药学会药物化学专委会委员,西安高新区首批签约创业导师。2013年于中国药科大学取得药物化学专业博士学位,2016年12月赴美国佐治亚州立大学王炳和教授课题组访问学习。长期以来从事小分子激酶抑制剂及抗菌药物研发、天然产物改造与生物活性研究。以第一作者或通信作者在 Journal of Medicinal Chemistry, European Journal of Medicinal Chemistry, Food & Function, Journal of Functional Foods, Biomedicine & Pharmacotherapy 和《中国抗生素杂志》等期刊发表 SCI、EI 收录论文 30 余篇,授权中国、美国及日本发明专利 60 余件,多数已在企业转化。